7 Aplicaciones de la Paleo-Virología.
La historia co-evolutiva entre retrovirus endógenos y su huésped humano refleja el patrón de migración de nuestra especie.
Los humanos somos seres a los que nos gusta clasificar. Y nos gusta mucho más clasificarnos a nosotros mismos; así pues, tenemos nombres, apellidos, familias, tribus, nacionalidades, etc. Este afán por clasificar también está unido a una necesidad de conocer de dónde venimos, de cuál fue el recorrido de nuestros ancestros para llegar a donde estamos hoy en día ([34]). Tradicionalmente estos estudios se hacen siguiendo registros de fósiles humanos, análisis arqueológicos, históricos, antropomórficos, socio-culturales y biogeográficos ([34]).
Sin embargo, con el advenimiento de la era genómica esto ha cambiado radicalmente ([35], [36], [37]). Nuestra capacidad para determinar las relaciones entre individuos, poblaciones y especies se está transformando gracias a las colecciones en crecimiento, actualmente con miles de genomas obtenidos a partir de ADN antiguo, bases de datos de muestras de interés médico, a escala poblacional, sumado a los esfuerzos para secuenciar millones de especies eucariotas. Estas relaciones, y las distribuciones de la variación genética y fenotípica resultante, reflejan el complejo conjunto de procesos y acontecimientos selectivos, demográficos y moleculares que han dado forma a las especies y, por consiguiente, son una rica fuente de información sobre ellas ([35], [36]).
Recientemente, los métodos para lograr inferir estas relaciones poblacionales en el humano han presentado numerosos retos debido al volumen de información disponible y el reto que implica depurarla ([36]). Estos estudios de genómica comparativa tienen el problema de poder llegar a ser costosos por muestra analizada, lo que restringe el número de muestras a utilizar, lo que conlleva a problemas de representación de las diversas etnias de la especie, y además, pueden dar resultados distintos, dependiendo de las regiones del genoma que se comparen o el método de estudio utilizados (SNLP, RFLP, ARN16s, etc.) ([35], [36]). Pero en particular, la variabilidad genética de estos marcadores limita la resolución temporal a evaluar, en otras palabras, la tasa de mutación en estas secuencias humanas es mucho más lenta que la tasa de migración.
Debido a lo anterior expuesto, nuevos y mejores métodos para indagar sobre nuestra biogeografía se encuentran en desarrollo. Uno de los métodos novedosos propuestos es la utilización de secuencias del viroma humano, es decir retro-virus endógenos activos, cuya mutación, al ser mucho mayor que las secuencias no-virales del genoma humano, permiten indagar escalas temporales cercanas a las empleadas en el estudio de las migraciones humanas ([35]).
Los virus representan buenos candidatos para estos estudios debido a la íntima co-evolución que se da entre el virus y el huésped, donde una carrera de competencia (arms race) entre factores virulentos del virus y la respuesta del huésped moldean eventos de introducción, expulsión del genoma, eventos evolutivos que nos permiten calibrar relojes moleculares y otros aspectos de la biología de ambos organismos, principalmente a través del análisis de eventos de recombinación ([35]).
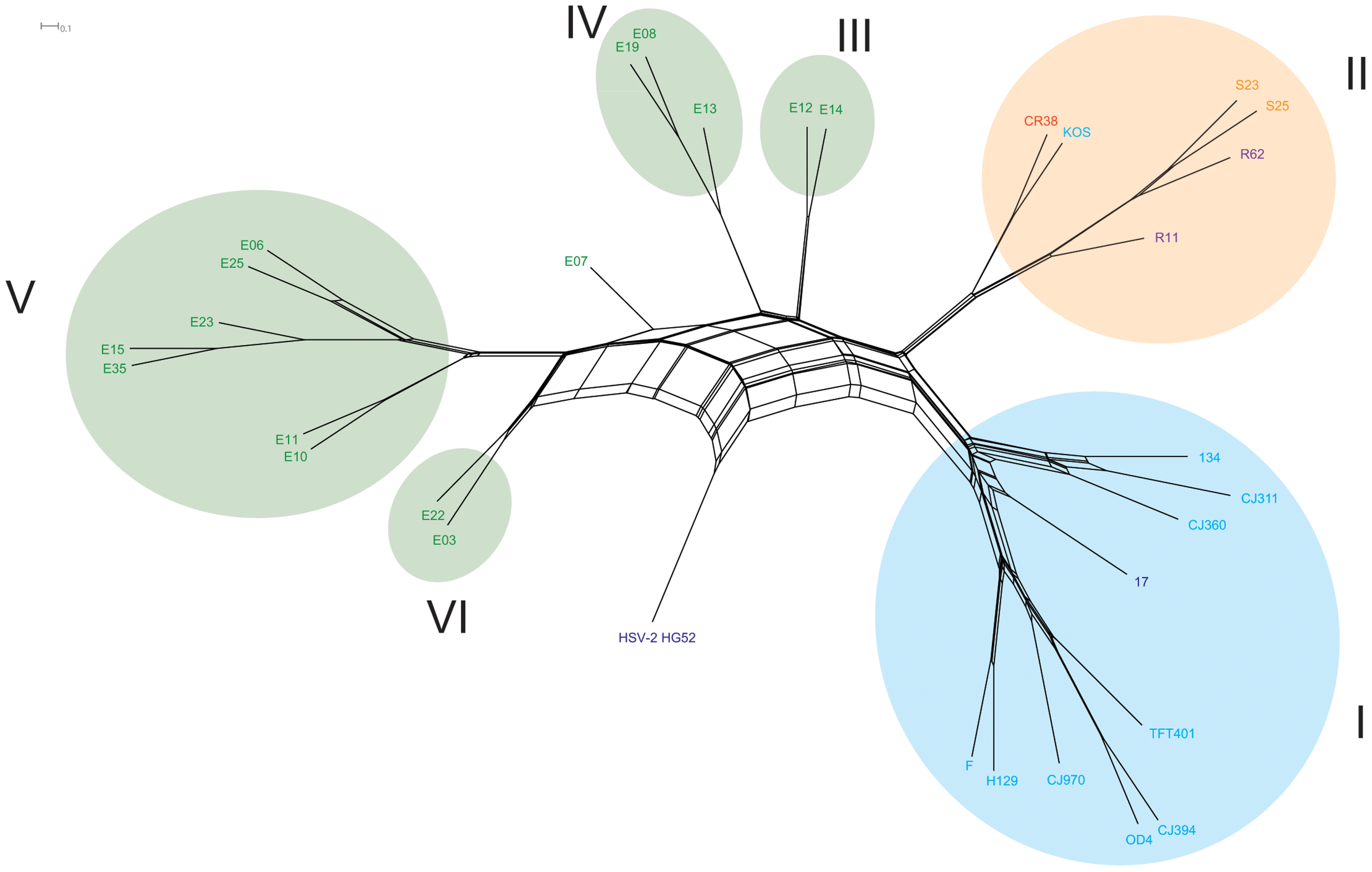
Figura 7.1: Árbol de secuencias consenso construido de una partición de 5kb del genoma del virus examinado mediante un análisis boostrap de máxima similitud con 500 repeticiones. Un consenso del 70% se usó como valor umbral para realizar las agrupaciones. El clado I corresponde a poblaciones norteamericanas y europeas , el clado II americanas y del asía oriental y los clados IIIV V y VI son de áfrica del éste. HSPV-2 se utilizó como grupo comparativo de afuera, tomado de [35]