7.1 Co-diversidficación virus del herpes simple y poblaciones humanas.
Los virus humanos siendo organismos de fácil secuenciamiento, en términos de la diminuta escala del genoma, y por lo general altamente conservados, este hecho ha permitido el desarrollo de una nueva especialidad en la virología evolutiva denominada paleo-virología, la cual, a partir del estudio de la coevolución huésped-parásito, se aproxima al entendimiento de eventos históricos de las especies, como la migración de las poblaciones humanas desde el lugar de origen, hasta el alcance de la distribución mundial.
El virus del herpes simple (VHS-1) es un candidato que ha mostrado ser ideal para este tipo de análisis. Los herpesvirus son grandes virus de ADN de doble cadena con genomas que varían en tamaño de 124 a 295 kilobases. La subfamilia de los alfaherpesvirus se caracteriza por la capacidad de establecer infecciones latentes en los ganglios nerviosos sensoriales. Estudios filogenéticos anteriores han demostrado que los herpesvirus han coevolucionado con sus huéspedes. El virus del herpes simple tipo 1 (HSV-1) es un alfaherpesvirus con un genoma de aproximadamente 152 Kb. El VHS-1 causa lesiones mucocutáneas orales, así como queratitis y encefalitis, y es un importante patógeno humano ([35])
Mediante análisis filogenéticos y frecuencias de recombinación se ha determinado que el VHS-1 presenta una estructura mínima de seis clados, cada uno correlacionado con regiones geográficas distintas. Los datos filogenéticos de las tasas de sustitución del VHS-1 sugiere una tasa de aproximadamente 1,38*10^7 subs/sitio/año. El análisis de recombinación del VHS-1 muestra evidencias de recombinación inter e intracladística (es decir recombinación dentro del mismo linaje) ([35]).
Estas características del virus se han utilizado en un muestreo global de cepas de VHS-1 para el análisis filogenético y apoya la conclusión de que las cepas de VHS-1 han co-migrado con sus huéspedes humanos, dando lugar a clados virales geográficamente separados ([35]). En la figura 2 se puede observar como la historia evolutiva de cada clado de VHS-1 correlaciona geográfica y temporalmente, con los eventos migratorios y de separación geográfica de los grandes grupos de poblaciones humanas existentes a lo largo del tiempo ([35]).
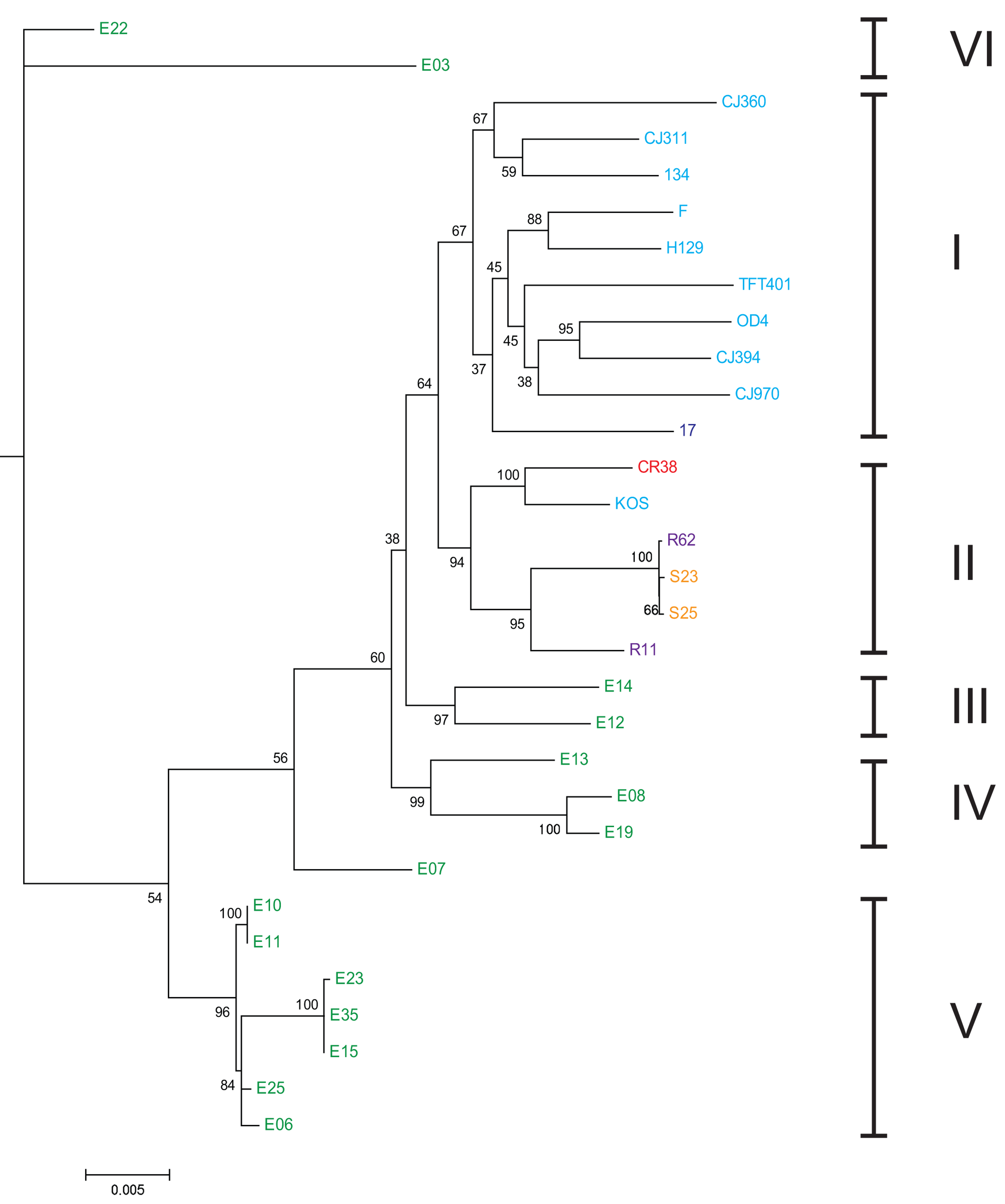
Figura 7.2: Árbol filogenético ML enraizado (HSV-2 se uso como grupo externo) de las distintas variedades de VHS-1 y su correlación con los 6 grupos de poblaciones humanas establecidas en en estudio (números latinos a la derecha). Nótese como cada clado corresponde a una población en particular, evidenciando una co-evolución del virus con cada población humana. De allí que el análisis de diversidad genética de HSV-2 resulta en una herramienta de análisis de migración y biogeografía humana. Los aislados virales de poblaciones actuales están coloreados según el país de origen: EE.UU.: azul claro, Reino Unido: azul oscuro, China: rojo, Corea del Sur: púrpura, Japón: amarillo y Kenia: verde, tomado de [35]